
歐洲
開始於1990年代,歐盟(EU)醫療器材指令持續性的發布關於醫療器材的相關規範,除了歐盟成員國外,其他各國如澳洲、瑞士、挪威等也都以歐盟標準作為訂定國家醫療器材法規的參考。經由全球醫材法規調和會(IMDRF)的努力,醫療器材指令可以用來協調各國在相關問題上的參考,歐盟近期的以下列指令包含了在醫療器材分類、產品認證方式、品質系統的要求以及技術文件上的規範::
歐盟執行委員會將醫療器材歸類為第一級、第二a級、第二b級、第三級基於風險等級,較高等級的醫療器材應該遵從較嚴苛的審查評估以取得CE標章。CE標章為歐盟法律規定任何器械、電器、以及其他許多類型產品要在歐盟地區上市前所需的認證。此標章代表產品經過歐盟認可,之後可以進行販賣。
有兩個方式可取得醫療器材的CE Marking,其一為經由歐盟核准機構取得認證,其二為符合標準聲明由廠商自行對產品聲稱他們的產品合乎安全規範。第二個方式只能適用在低風險的醫療器材,高風險產品必須經由第一種方式。不同於美國食品藥品監督管理局,歐盟執行委員會要求在申請CE Marking時需要有品質系統的驗,品質系統應該要遵守相關的醫療器材法規來建置安全品質系統。在CE Marking申請時,申請人需要提交技術文件(TCF)以提供科學證據和檢驗資料來佐證醫療器材。
根據規定非歐盟會員國的外國廠商必須要指定歐體代表來進行CE Marking申請,歐體代表需要是在歐洲經濟區內有註冊地址的自然人或法人團體。歐體代表的功用就是作為外國廠商與歐盟主管機關溝通的窗口並且要負責提交申請文件。在歐盟範圍內的政府與相關主管機關有權在任意時機聯絡歐體代表來了解是否海外廠商有符合相關規範。弘亞生技顧問有限公司輔導客戶CE Marking申請、品質系統建置、歐體代表還有各種進入歐洲市場所需的申請流程。歡迎聯絡我們詢問任何關於進入歐盟醫療器材市場的法規問題。
- 主動式植入醫療器材 AIMD, 90/76/EC
- 醫療器材指令MDD, 93/42/EEC
- 體外檢測醫療器材 IVDD, 98/79/EC
歐盟執行委員會將醫療器材歸類為第一級、第二a級、第二b級、第三級基於風險等級,較高等級的醫療器材應該遵從較嚴苛的審查評估以取得CE標章。CE標章為歐盟法律規定任何器械、電器、以及其他許多類型產品要在歐盟地區上市前所需的認證。此標章代表產品經過歐盟認可,之後可以進行販賣。
有兩個方式可取得醫療器材的CE Marking,其一為經由歐盟核准機構取得認證,其二為符合標準聲明由廠商自行對產品聲稱他們的產品合乎安全規範。第二個方式只能適用在低風險的醫療器材,高風險產品必須經由第一種方式。不同於美國食品藥品監督管理局,歐盟執行委員會要求在申請CE Marking時需要有品質系統的驗,品質系統應該要遵守相關的醫療器材法規來建置安全品質系統。在CE Marking申請時,申請人需要提交技術文件(TCF)以提供科學證據和檢驗資料來佐證醫療器材。
根據規定非歐盟會員國的外國廠商必須要指定歐體代表來進行CE Marking申請,歐體代表需要是在歐洲經濟區內有註冊地址的自然人或法人團體。歐體代表的功用就是作為外國廠商與歐盟主管機關溝通的窗口並且要負責提交申請文件。在歐盟範圍內的政府與相關主管機關有權在任意時機聯絡歐體代表來了解是否海外廠商有符合相關規範。弘亞生技顧問有限公司輔導客戶CE Marking申請、品質系統建置、歐體代表還有各種進入歐洲市場所需的申請流程。歡迎聯絡我們詢問任何關於進入歐盟醫療器材市場的法規問題。
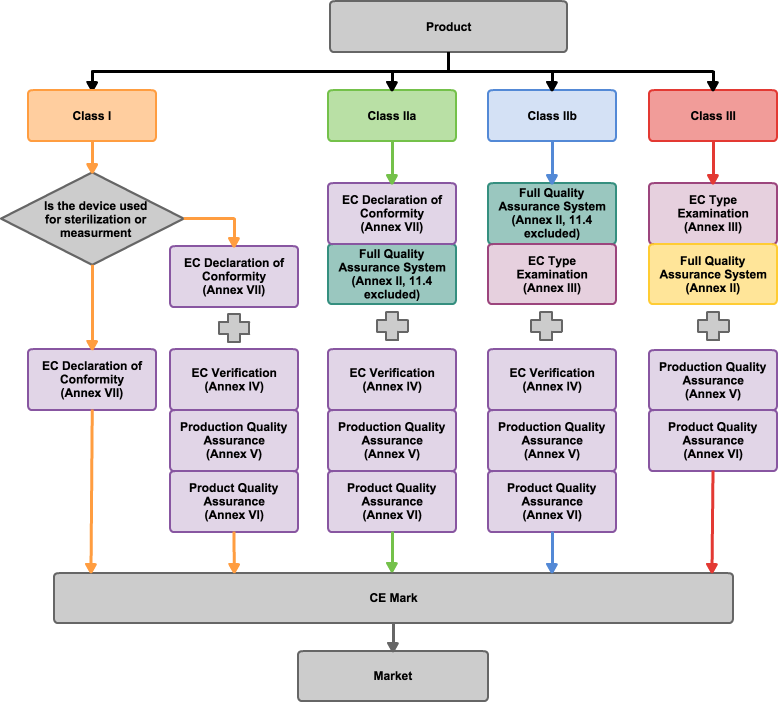
CE Marking流程圖
